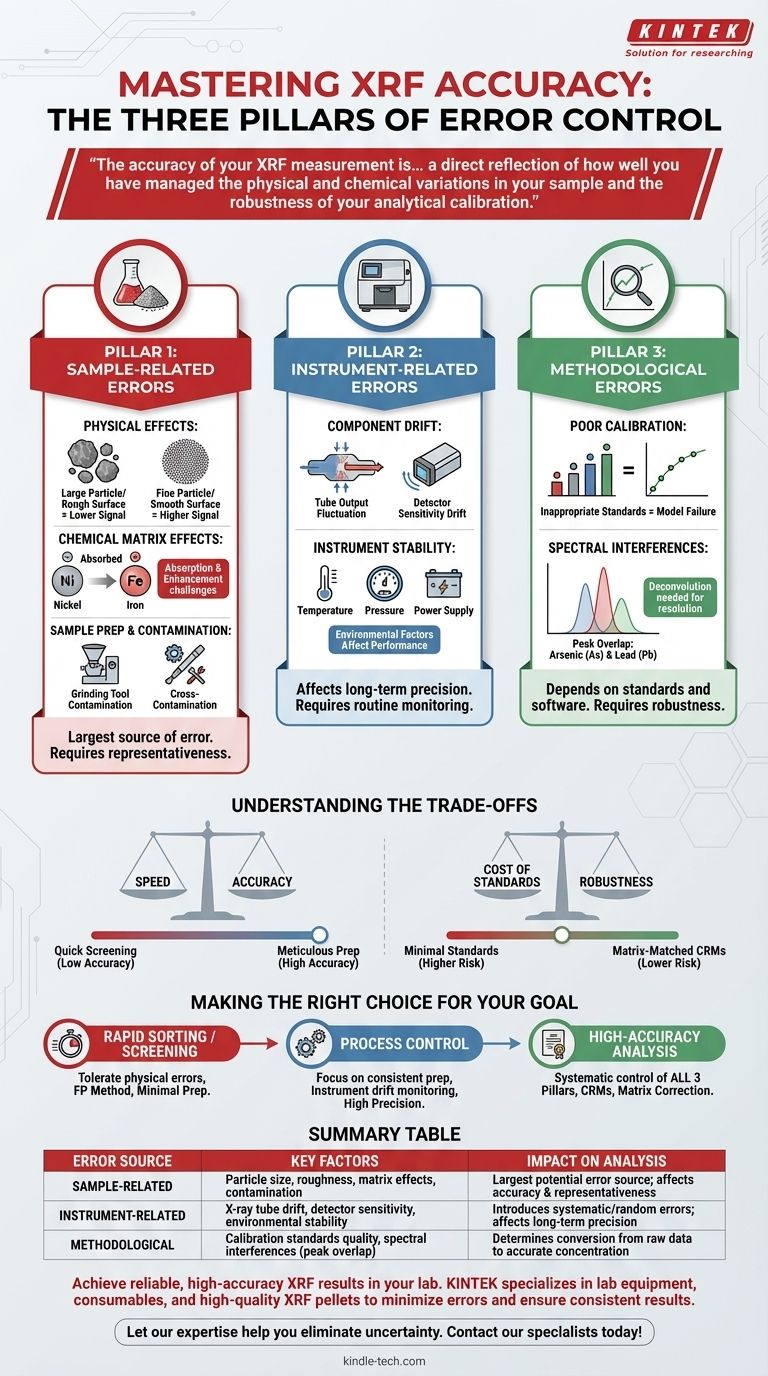
In X-ray Fluorescence (XRF) analysis, errors originate from three primary domains: the sample itself (physical and chemical properties), the instrument's hardware and stability, and the analytical method or calibration being used. While factors like sample contamination during preparation are significant, a truly accurate result depends on controlling variables across all three areas.
The accuracy of your XRF measurement is not just a function of the spectrometer's quality, but a direct reflection of how well you have managed the physical and chemical variations in your sample and the robustness of your analytical calibration.

The Three Pillars of XRF Error
To achieve reliable results, you must understand where potential inaccuracies can be introduced. We can group these sources into three distinct categories.
Pillar 1: Sample-Related Errors
This is often the largest source of error. The spectrometer can only measure the sample it is presented with; if the sample isn't a true representation of the bulk material, the results will be flawed.
Physical Effects
The physical nature of the sample dramatically influences the X-ray signal. Key factors include particle size, surface roughness, and sample uniformity.
Finer particles generally produce a more intense fluorescent signal than larger ones. An inconsistent grind or a rough surface can lead to significant, unpredictable errors.
Chemical Matrix Effects
This refers to how other elements in the sample absorb or enhance the X-rays from the element you are trying to measure. This is a fundamental challenge in XRF.
For example, a high concentration of iron will heavily absorb the fluorescence from nickel, making the nickel appear less concentrated than it truly is. These effects must be corrected mathematically.
Sample Preparation and Contamination
The way a sample is prepared is a critical control point. Errors introduced here are irreversible.
As noted, contamination from grinding equipment can introduce foreign elements. Similarly, sample-to-sample cross-contamination can occur if preparation tools are not meticulously cleaned between uses.
Pillar 2: Instrument-Related Errors
While modern XRF spectrometers are highly stable, they are not perfect. Hardware variations can introduce systematic or random errors into the analysis.
Component Drift
The two most critical components, the X-ray tube and the detector, can experience performance changes over time.
The tube's output intensity can fluctuate, and the detector's sensitivity can drift due to temperature changes or aging. These changes are usually slow and can be managed with routine monitoring.
Instrument Stability
Factors like ambient temperature, barometric pressure (in some systems), and power supply stability can affect the spectrometer's performance.
Maintaining a controlled environment for the instrument is crucial for achieving high-precision, long-term analytical results.
Pillar 3: Methodological and Calibration Errors
Even with a perfect sample and a stable instrument, the final result depends entirely on the analytical method and the quality of the calibration.
Poor Calibration
The calibration is the mathematical model that converts raw X-ray intensities into element concentrations. This model is only as good as the standards used to create it.
Using insufficient or inappropriate calibration standards that don't match the chemical matrix of your unknown samples is a primary source of major analytical error.
Spectral Interferences
Sometimes, the characteristic X-ray lines of two different elements are so close together that the detector cannot resolve them. This is known as peak overlap.
For example, the K-alpha line of arsenic overlaps with the K-beta line of lead. Sophisticated software is needed to mathematically deconvolve these peaks and report an accurate result for each element.
Understanding the Trade-offs
Controlling every source of error can be time-consuming and expensive. The key is to match your preparation and analytical rigor to your specific goal.
Speed vs. Accuracy
A quick, "point-and-shoot" analysis on an unprepared sample might be sufficient for simple material identification or screening.
However, this approach sacrifices accuracy and is completely unsuitable for quality control or regulatory compliance, where meticulous sample preparation (like grinding and pressing pellets) is non-negotiable.
Cost of Standards vs. Robustness
Creating a robust calibration requires a wide range of high-quality, matrix-matched certified reference materials, which can be expensive.
Using a minimal set of standards or relying on "type-standardization" (adjusting a factory calibration with one or two local samples) is cheaper but introduces a higher risk of error if your samples deviate from the standards.
Making the Right Choice for Your Goal
Your analytical strategy should be dictated by the question you need to answer.
- If your primary focus is rapid material sorting or screening: You can often tolerate errors from physical effects and use a simple fundamental parameters (FP) method, minimizing sample preparation.
- If your primary focus is process control with a known material type: Your main concern is precision, so focus on highly consistent sample preparation and routine instrument drift monitoring.
- If your primary focus is high-accuracy analysis for certification or research: You must systematically address all three pillars, using meticulous sample preparation, certified reference materials for calibration, and matrix correction software.
Ultimately, achieving accuracy in XRF is an exercise in systematic control, where understanding the potential sources of error is the first step toward eliminating them.
Summary Table:
| Error Source | Key Factors | Impact on Analysis |
|---|---|---|
| Sample-Related | Particle size, surface roughness, chemical matrix effects, contamination | Largest potential source of error; affects result accuracy and representativeness |
| Instrument-Related | X-ray tube drift, detector sensitivity, environmental stability | Introduces systematic or random errors; affects long-term precision |
| Methodological | Calibration standards quality, spectral interferences (peak overlap) | Determines the conversion from raw data to accurate concentration values |
Achieve reliable, high-accuracy XRF results in your lab.
The path to precise analysis requires controlling variables across your sample, instrument, and method. KINTEK specializes in lab equipment and consumables, serving laboratory needs with high-quality XRF pellets, presses, and accessories designed to minimize sample preparation errors and ensure consistent results.
Let our expertise help you eliminate uncertainty. Contact our specialists today to discuss your specific application and how we can support your analytical goals.
Visual Guide